
27
Sep
Generate a genome index using genome reference information. STAR outputs read counts per gene into PREFIXReadsPerGeneouttab file with 4 columns which correspond to different strandedness options.
Star rna seq aligner. STAR can be installed on FreeBSD via the FreeBSD ports system. To install via the binary package simply run. Pkg install star LIMITATIONS.
This release was tested with the default. STAR is an aligner designed to specifically address many of the challenges of RNA-seq data mapping using a strategy to account for spliced alignments. STAR Alignment Strategy STAR is shown to have high accuracy and outperforms other aligners by more than a factor of 50 in mapping speed but it is memory intensive.
In addition to detecting annotated and novel splice junctions STAR is capable of discovering more complex RNA sequence arrangements such as chimeric and circular RNA. STAR can align spliced sequences of any length with moderate error rates providing scalability for. Spliced Transcripts Alignment to a Reference STAR is a fast RNA-seq read mapper with support for splice-junction and fusion read detection.
STAR aligns reads by finding the Maximal Mappable Prefix MMP hits between reads or read pairs and the genome using a Suffix Array index. Different parts of a read can be mapped to different. STAR is an aligner designed to specifically address many of the challenges of RNA-seq data mapping using a strategy to account for spliced alignments.
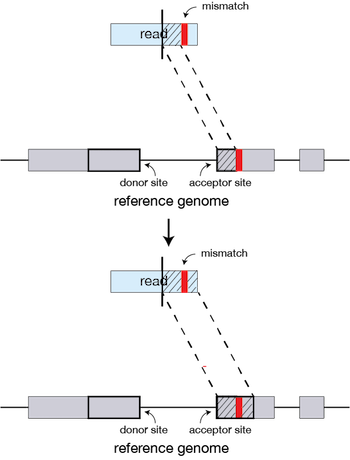
We will not go into detail about how STAR works but if you are interested in undertanding the alignment strategy we have some materials linked here. In this step user supplies the genome les generated in the 1st step as well as the RNA-seq reads sequences in the form of FASTA or FASTQ les. STAR maps the reads to the genome and writes several output les such as alignments SAMBAM mapping summary statistics.
STAR outputs read counts per gene into PREFIXReadsPerGeneouttab file with 4 columns which correspond to different strandedness options. Gene ID column 2. Counts for unstranded RNA-seq column 3.
Counts for the 1st read strand aligned with RNA htseq-count option -s yes column 4. Counts for the 2nd read strand aligned with RNA htseq-count option -s reverse. Contribute to alexdobinSTAR development by creating an account on GitHub.
STAR command line has the following format. STAR –option1-name option1-values–option2-name option2-values. If an option can accept multiple values they are separated by spaces and in a few cases - by commas.
2 Generating genome indexes. The basic options to generate genome indices are as follows–runThreadN NumberOfThreads. Arriba is a fusion detection algorithm based on the STAR RNA-Seq aligner.
It is the winner of the DREAM Challenge about fusion detection. 79 Arriba can also detect viral integration sites internal tandem duplications whole exon duplications circular RNAs enhancer hijacking events involving immunoglobulinT-cell receptor loci and. STAR is a fast RNA-Seq read mapper with support for splice-junction and fusion read detection and it was designed to align non-contiguous sequences directly to a reference genome.
STAR aligns reads by finding maximal mappable prefix hits between reads or read pairs and the genome using a. Align expression data with STAR. To align the RNA transcripts to the reference genome we will make use of STAR 2.
Alignment with STAR is a two-step process. Generate a genome index using genome reference information. Align sequencing data using the.
Read alignment quality control and data analysis were performed using Visualization Pipeline for RNA-seq VIPER 67. Sequencing reads were aligned using STAR read alignment 68. STAR is an aligner designed to specifically address many of the challenges of RNA-seq data mapping using a strategy to account for spliced alignments.
STAR is shown to have high accuracy and outperforms other aligners by more than a factor of 50 in mapping speed but it is memory intensive. BioCloud RNA-Seq STAR Result Documentation. The first step of Cufflinks is to assemble all possible transcripts with the results of alignment from TopHat.
To accurately estimate the abundance of each transcript any possible isoforms are considered in this step. Then Cuffmerge merges the previous assembles of individual condition. Aligning RNA-seq data The theory behind aligning RNA sequence data is essentially the same as discussed earlier in the book with one caveat.
RNA sequences do not contain introns. Gene models in Eukaryotes contain introns which are often spliced out during transcription.
Previous post
State of the art research proposalNext post
Stages of power supply